Welp, I've shifted my focus from general health to the way gut health affects overall health. Tons of new info you won't find anywhere else. I think I'm writing it from a much different perspective than other bloggers. I'm more interested in how the environment of the gut is regulated and what we can do to tip the scales in our favor.
You can find my new blog with more articles at www.hackyourgut.com. You can also request to join my private facebook group here.
Thanks for reading here, there, or anywhere.
Dave
Friday, March 24, 2017
Thursday, May 19, 2016
Detoxification 101: The basic science behind cellular detoxification
Are you sensitive to mold, perfumes, detergents, or other chemicals in the environment? Do you suffer from a hormonal imbalance that includes the thyroid, adrenal, and sex hormones? Have you attempted to rectify the situation by performing a detox, cleanse, or other form of reset with little or no improvement? Well maybe it's time you learned the finer points of detoxification. In this blog we'll cover:
Once your finished with this blog, you'll have a pretty good handle on the detoxification process and how you can use it to your advantage to improve your health. You'll also understand how certain supplements or foods that are considered detox powerhouses may not be right for every person. An individualized approach is best, and sometimes throwing random supplements and foods at the problem can actually make you feel worse. Let's jump in.
Along with looking different, each cell type in your body contains different functional elements. From a detoxification perspective, we are going to focus on something called the steroid and xenobiotic receptor, or SXR. As the name suggests, the SXR is a receptor that binds steroid hormones and xenobiotics. Xenobiotics are simply molecules that are foreign to the body.
Something that binds to a receptor is called a ligand. When a ligand binds to the SXR, it initiates the process of biotransformation. For a steroid hormone, this means that it recruits enzymes that either activate or deactivate that steroid hormone. For a xenobiotic, binding to the SXR initiates the 3 phases of detoxification for eventual removal, but we'll get back to that. What's important to understand is that cells that express high levels of this receptor will be highly involved in the detoxification process, and the process is exactly the same whether you are transforming a steroid hormone, detoxifying a xenobiotic, or metabolizing a pharmaceutical drug.
When we look at cells that express high levels of the SXR, we find 2 separate functional groups. The first is organs involved in the manufacture and transformation of steroid hormones which include the liver, kidney/adrenals, and reproductive organs. The second functional group is involved in the detoxification of xenobiotic substances and include the lung, skin, digestive tract, liver, and kidneys/adrenals. For the moment we'll focus on detoxification, but we'll swing back to the steroid part in a little bit.

Of course, no process is 100% efficient, and the GI tract is no exception. Whether it be from sluggish detoxification or an impairment in the intestinal barrier, xenobiotics can gain entrance from time to time. Fortunately, xenobiotics that enter the circulation from the GI tract do so through the portal vein, which immediately goes to the liver for further detoxification. This doesn't mean that you can't overwhelm the system, and there are numerous xenobiotics that enter the digestive tract on a daily basis. The ones you need to be primarily concerned with are fat soluble. This includes what is likely the most ubiquitous xenobiotic to enter the human digestive tract, bisphenol-A(BPA).
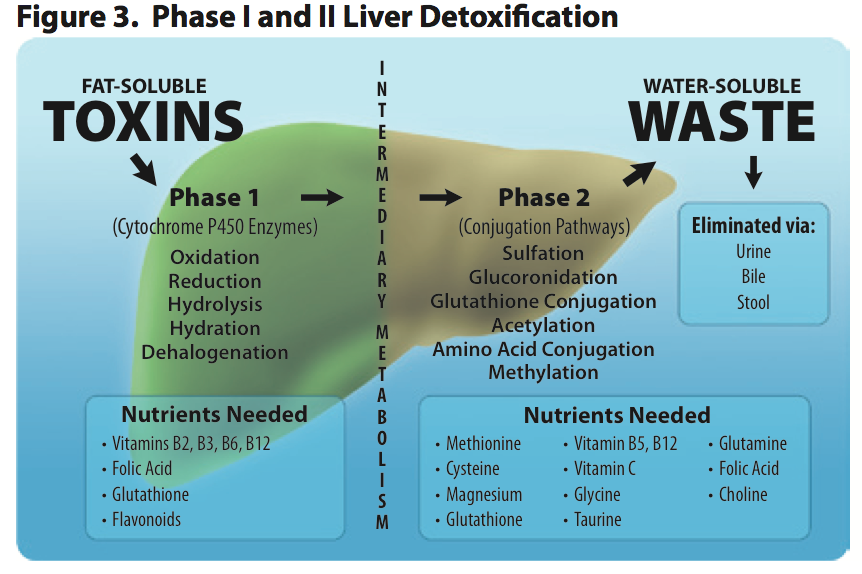
This brings us to fat soluble toxins. As the name suggests, fat soluble toxins can freely enter your cells because they are fat soluble and the plasma membrane is made of fat. When a fat soluble toxin enters one of your cells, the SXR binds to the toxin in the cytoplasm and initiates one or all of the 3 phases of detoxification. The 3 phases of detoxification include modification, conjugation, and transport/excretion.
Phase 1 detoxification modifies the fat soluble toxin through either oxidation, reduction, or hydrolysis to make the toxin more water soluble. This is typically done by one of the cytochrome P450 family of enzymes, of which there are 18. In some instances this makes the toxin less toxic and easy to remove from the cell and the body. However, there are instances where Phase 1 detoxification isn't enough to remove the toxin from the cell, and sometimes Phase 1 actually makes the toxin more toxic. A great example of this is the conversion of alcohol to acetaldehyde, which is more toxic than alcohol.
If the toxin cannot be removed by the cell or is made more toxic during Phase 1, Phase 2 detoxification begins. This further transforms the toxin so that it's readily soluble in water and can be removed from the cell. Phase 2 conjugation is carried out through 6 different pathways including methylation, sulphation, acetylation, glucuronidation, glutathione conjugation, and glycine conjugation. Once the offending molecule is conjugated, Phase 3 transporters escort the now water soluble toxin out of the cell. Since it's now water soluble, it can't enter your cells and is excreted via the urine, sweat, or dumped in to the feces as a component of bile, which is produced by the liver.

Any fat soluble toxin that makes it past the GI tract and liver, or that enters through one of the other barriers, is capable of interacting with the SXR in any cell that expresses it. This is problematic in organs involved in the transformation of hormones to active or inactive states including the above mentioned liver, adrenals, and reproductive organs. When a toxin that is a ligand for SXR enters the circulation, it can bind to the SXR in any tissue and disrupt signaling there.
Probably the best known fat soluble toxin that is also a ligand for the SXR is bisphenol-A or BPA. BPA is an endocrine-disrupting chemical capable of binding multiple types of hormone receptors, the SXR being one of them. BPA typically enters the circulation through the GI tract. However, under normal circumstances, BPA enters the general circulation in its inactive, conjugated form after undergoing phase 2 detoxification in the GI tract and/or liver.
Any BPA that enters the general circulation in the conjugated form will not enter tissues because it's water soluble and has no transporter to bring it across cell membranes. This makes it readily excreted from the body. If BPA enters the circulation in its active, unconjugated form, it can enter cells, bind to the SXR, and disrupt the biotransformation of hormones in steroidogenic tissues. In the reproductive organs, this will lead to fertility issues. In the adrenal glands, SXR is primarily involved in corticosterone and aldosterone production. This would lead to the problems associated with HPA axis dysfunction which are primarily due to electrolyte imbalance
Corticosterone is also of particular interest in the skin as it helps regulate inflammation there. The skin has its own functioning HPA axis similar to the HPA axis that controls cortisol secretion from the adrenals, but the final product in the skin is corticosterone. In the skin, corticotropin releasing factor(CRF) causes the secretion of adrenocorticotropic hormone(ACTH) which then causes the release of corticosterone. Corticosterone then feeds back to stop the release of CRF. Unregulated CRF released causes inflammation in the skin by causing mast cells to release histamine, which is why topical hydrocortisone is used to treat skin inflammation: it stops CRF release via negative feedback due to its structural similarity to corticosterone.
This is all well and good, but the questions remains...How would unconjugated BPA get past the intestinal barrier and liver?

It's well known that people with inflammatory bowel disease have impaired detoxification. The primary reason for this is because the SXR and inflammation are involved in reverse regulation of one another. Specifically, when inflammation goes up the expression of the SXR drops and when SXR is activated inflammation levels drop. We know this both through direct evidence from scientific studies as well as the effect of systemic inflammation on pharmaceutical drug metabolism. Approximately 60% of pharmaceutical drugs on the market today are metabolized by cytochrome p450 3A4(CYP3A4) which is controlled by the SXR, and why wouldn't they be? Many pharmaceutical drugs are steroids, all are xenobiotics, and a good number of them could be considered both.
So the answer to how unconjugated BPA can get past the intestinal barrier and liver is via inflammation. Inflammation in the digestive tract impairs detoxification there and increases intestinal permeability. This allows unconjugated BPA to enter the portal vein while systemic inflammation impairs the livers ability to conjugate BPA once it makes its way there. The result...unconjugated BPA accesses the general circulation where it can disrupt hormonal signaling throughout the body.
(Quick sidebar: a few years ago the scientific consensus was that BPA was so quickly metabolized that unconjugated BPA never entered the general circulation, despite multiple lines of evidence to the contrary. Of course, the studies that conflicted with industry studies were disregarded as being due to laboratory error or contamination. At this point, the science is overwhelming. The current understanding is that either BPA is not detoxified as rapidly as once thought, that there are other routes outside the digestive tract that allow unconjugated BPA in, or the most likely answer being both. Regardless, it's entering the circulation unconjugated and disrupting hormonal signaling)

The next thing you can do is limit the amount of fat soluble xenobiotics you are exposed to on a daily basis. BPA is a big one, primarily due to the consumption of food heated in plastic, cooked with plastic utensils, or hot coffee that's exposed to plastic. This includes coffee made in coffeemakers that use plastic including Keurig brewers, coffee that's stored in plastic dispensers at gas stations and convenience stores, and coffee that's consumed through plastic lids. Whether these containers are BPA-free or not is irrelevant, the most frequently used replacement for BPA, BPS, has the same endocrine disrupting effects as BPA and seeps from plastic container in to its contents. Organochlorine pesticides are also a major route of exposure. Other sources of fat soluble toxins include receipts, cosmetics, detergents and perfumes, although these sources shouldn't be ingested but may enter via the skin. :)
One thing you shouldn't do is just start slamming down detoxification promoting supplements and foods. Manipulating the detoxification process to improve your health is a highly individualized process dependent on genetic as well as environmental factors and many substances can have a dramatic impact on the way you feel due to these factors. For example, grapefruit and pomegranate juice are both known to inhibit phase 1 detoxification and can dramatically impact the blood levels of certain drugs, actually a lot of them since they both directly inhibit CYP3A4. So think about that when your slamming down a bottle of Pom. Also, feel free to refer this to your friend who says the human body can easily handle all environmental toxins that are thrown at it when simply drinking grapefruit juice can lead to toxic accumulation of pharmaceutical drugs in a very short amount of time.
For someone on chemotherapy or a drug metabolized via CYP3A4, these products can lead to adverse events. However, for someone like me who is genetically a fast oxidizer, slowing down phase 1 detoxification can reduce the number of products from phase 1 detoxification and allow my phase 2 pathways to catch up, improving fatigue and general well-being due to reduced toxic load.
Another area where Phase 1 inhibitors are useful is people reacting to aflatoxins from mold. Alflatoxins are made more toxic after Phase 1 reactions and can bioaccumulate if Phase 2 is outpaced by Phase 1. Different supplements manipulate detoxification in different ways and this needs to be kept in mind when supplementing to improve the way you feel as well as your overall health.
This brings me to a final point...There are 6 pathways of phase 2 detoxification, don't just focus on methylation and glutathione conjugation. BPA is a great example given that it's conjugated primarily via glucuronidation and sulphation, 2 pathways that use and are induced by different nutrients than methylation and glutathione conjugation. Supporting these two pathways doesn't do too much for the removal of BPA from the body. In fact, simply sweating does more for BPA removal than supporting methylation and glutathione conjugation, although glutathione can act as a back up if the other 2 pathways fail.
Fortunately, the absolute first step is relatively easy to do: reduce inflammation. This is primarily done through lifestyle modification including diet, exercise, stress management, proper sleep, and other lifestyle factors that are associated with inflammation. People with inflammatory conditions such as IBD/IBS, arthritis, or an active autoimmune condition will almost certainly require some other support which can include pharmaceutical drugs, supplements, or a combination of both.
From there, if you want to work your detoxification pathways, the next step is knowing how detoxification works for your specific issues. Having your genome done by 23andme and using promethease or genetic genie to look at your detoxification pathways is very useful and can help you identify dietary and supplemental factors that will save you a lot of time and failure in the long run. It will help you identify which pathways are sluggish and, therefore, which environmental toxins you may be more sensitive to. There are numerous resources on the web that can get you pointed in the right direction.
The final step is to formulate a plan and give it a go, using how you feel as a guide on your success and recording what you do for future reference. Also, keep in mind that initiating any program like this should be discussed with your physician so that any drug-drug and drug-supplement interactions can be avoided.
- The layout of the human body from a detox perspective
- The 3 phases of detoxification at the cellular level
- The hormone/detox link
- How inflammation blocks detoxification
- Other factors that likely impede your ability to detox
Once your finished with this blog, you'll have a pretty good handle on the detoxification process and how you can use it to your advantage to improve your health. You'll also understand how certain supplements or foods that are considered detox powerhouses may not be right for every person. An individualized approach is best, and sometimes throwing random supplements and foods at the problem can actually make you feel worse. Let's jump in.

Detoxification at the organ level
The layout of the human body is quite amazing. You have trillions of cells that all contain the same DNA but look vastly different. This is because the environment your cells are in dictate what they become through a process called epigenetics. So even though your skin and liver cells contain precisely the same information, the environment each cell type is in causes each to differentiate in to a completely different type of cell that suits that particular environment.Along with looking different, each cell type in your body contains different functional elements. From a detoxification perspective, we are going to focus on something called the steroid and xenobiotic receptor, or SXR. As the name suggests, the SXR is a receptor that binds steroid hormones and xenobiotics. Xenobiotics are simply molecules that are foreign to the body.
Something that binds to a receptor is called a ligand. When a ligand binds to the SXR, it initiates the process of biotransformation. For a steroid hormone, this means that it recruits enzymes that either activate or deactivate that steroid hormone. For a xenobiotic, binding to the SXR initiates the 3 phases of detoxification for eventual removal, but we'll get back to that. What's important to understand is that cells that express high levels of this receptor will be highly involved in the detoxification process, and the process is exactly the same whether you are transforming a steroid hormone, detoxifying a xenobiotic, or metabolizing a pharmaceutical drug.
When we look at cells that express high levels of the SXR, we find 2 separate functional groups. The first is organs involved in the manufacture and transformation of steroid hormones which include the liver, kidney/adrenals, and reproductive organs. The second functional group is involved in the detoxification of xenobiotic substances and include the lung, skin, digestive tract, liver, and kidneys/adrenals. For the moment we'll focus on detoxification, but we'll swing back to the steroid part in a little bit.
The layout of the human body from a detox perspective
From a detoxification perspective, the human body is laid out very well. The 3 primary barriers keeping the external environment from invading our internal environment are well suited to detoxify xenobiotics because they all express the SXR. Of the 3 barriers, the one that is most often exposed to xenobiotics is the gastrointestinal tract, and it has nearly the same level of the SXR as the liver which has the greatest expression of SXR and has a central role in detoxification.Of course, no process is 100% efficient, and the GI tract is no exception. Whether it be from sluggish detoxification or an impairment in the intestinal barrier, xenobiotics can gain entrance from time to time. Fortunately, xenobiotics that enter the circulation from the GI tract do so through the portal vein, which immediately goes to the liver for further detoxification. This doesn't mean that you can't overwhelm the system, and there are numerous xenobiotics that enter the digestive tract on a daily basis. The ones you need to be primarily concerned with are fat soluble. This includes what is likely the most ubiquitous xenobiotic to enter the human digestive tract, bisphenol-A(BPA).
Cellular detoxification
When it comes to detoxification at the cellular level, water soluble toxins are really not that much of a concern for a couple of reasons. First off, all of our cells are surrounded by a plasma membrane made up of fat. This prevents water soluble toxins from entering in to cells unless there is a transporter for them, and most transporters that bring water soluble molecules in to cells are highly specific to water soluble substances that are created by the body. The other reason why water soluble toxins aren't much of a concern is because they can be readily excreted in the urine, sweat, or feces.This brings us to fat soluble toxins. As the name suggests, fat soluble toxins can freely enter your cells because they are fat soluble and the plasma membrane is made of fat. When a fat soluble toxin enters one of your cells, the SXR binds to the toxin in the cytoplasm and initiates one or all of the 3 phases of detoxification. The 3 phases of detoxification include modification, conjugation, and transport/excretion.
Phase 1 detoxification modifies the fat soluble toxin through either oxidation, reduction, or hydrolysis to make the toxin more water soluble. This is typically done by one of the cytochrome P450 family of enzymes, of which there are 18. In some instances this makes the toxin less toxic and easy to remove from the cell and the body. However, there are instances where Phase 1 detoxification isn't enough to remove the toxin from the cell, and sometimes Phase 1 actually makes the toxin more toxic. A great example of this is the conversion of alcohol to acetaldehyde, which is more toxic than alcohol.
If the toxin cannot be removed by the cell or is made more toxic during Phase 1, Phase 2 detoxification begins. This further transforms the toxin so that it's readily soluble in water and can be removed from the cell. Phase 2 conjugation is carried out through 6 different pathways including methylation, sulphation, acetylation, glucuronidation, glutathione conjugation, and glycine conjugation. Once the offending molecule is conjugated, Phase 3 transporters escort the now water soluble toxin out of the cell. Since it's now water soluble, it can't enter your cells and is excreted via the urine, sweat, or dumped in to the feces as a component of bile, which is produced by the liver.
The hormone/detox link
As you may have guessed, something called the steroid and xenobiotic receptor is a major link between detoxification and issues with hormonal balance. Ideally, most fat soluble toxins will bind the SXR in cells of the GI tract and get detoxified, and any that bypass the GI tract are dealt with by the liver and either removed from the body through urine or re-deposited in the GI tract in the bile after being transformed in to a water soluble molecule that cannot re-enter the circulation. The problem is, things don't always go as planned.Any fat soluble toxin that makes it past the GI tract and liver, or that enters through one of the other barriers, is capable of interacting with the SXR in any cell that expresses it. This is problematic in organs involved in the transformation of hormones to active or inactive states including the above mentioned liver, adrenals, and reproductive organs. When a toxin that is a ligand for SXR enters the circulation, it can bind to the SXR in any tissue and disrupt signaling there.
Probably the best known fat soluble toxin that is also a ligand for the SXR is bisphenol-A or BPA. BPA is an endocrine-disrupting chemical capable of binding multiple types of hormone receptors, the SXR being one of them. BPA typically enters the circulation through the GI tract. However, under normal circumstances, BPA enters the general circulation in its inactive, conjugated form after undergoing phase 2 detoxification in the GI tract and/or liver.
Any BPA that enters the general circulation in the conjugated form will not enter tissues because it's water soluble and has no transporter to bring it across cell membranes. This makes it readily excreted from the body. If BPA enters the circulation in its active, unconjugated form, it can enter cells, bind to the SXR, and disrupt the biotransformation of hormones in steroidogenic tissues. In the reproductive organs, this will lead to fertility issues. In the adrenal glands, SXR is primarily involved in corticosterone and aldosterone production. This would lead to the problems associated with HPA axis dysfunction which are primarily due to electrolyte imbalance
Corticosterone is also of particular interest in the skin as it helps regulate inflammation there. The skin has its own functioning HPA axis similar to the HPA axis that controls cortisol secretion from the adrenals, but the final product in the skin is corticosterone. In the skin, corticotropin releasing factor(CRF) causes the secretion of adrenocorticotropic hormone(ACTH) which then causes the release of corticosterone. Corticosterone then feeds back to stop the release of CRF. Unregulated CRF released causes inflammation in the skin by causing mast cells to release histamine, which is why topical hydrocortisone is used to treat skin inflammation: it stops CRF release via negative feedback due to its structural similarity to corticosterone.
This is all well and good, but the questions remains...How would unconjugated BPA get past the intestinal barrier and liver?
Inflammation and detoxification
Inflammation seems to be the major buzzword in medical sciences these days, and for good reason. It seems to be one of the underlying causes of many of the disease that are killing us and it disrupts many biological processes in the human body. If you haven't figured it out yet, one of the processes it affects is detoxification.It's well known that people with inflammatory bowel disease have impaired detoxification. The primary reason for this is because the SXR and inflammation are involved in reverse regulation of one another. Specifically, when inflammation goes up the expression of the SXR drops and when SXR is activated inflammation levels drop. We know this both through direct evidence from scientific studies as well as the effect of systemic inflammation on pharmaceutical drug metabolism. Approximately 60% of pharmaceutical drugs on the market today are metabolized by cytochrome p450 3A4(CYP3A4) which is controlled by the SXR, and why wouldn't they be? Many pharmaceutical drugs are steroids, all are xenobiotics, and a good number of them could be considered both.
So the answer to how unconjugated BPA can get past the intestinal barrier and liver is via inflammation. Inflammation in the digestive tract impairs detoxification there and increases intestinal permeability. This allows unconjugated BPA to enter the portal vein while systemic inflammation impairs the livers ability to conjugate BPA once it makes its way there. The result...unconjugated BPA accesses the general circulation where it can disrupt hormonal signaling throughout the body.
(Quick sidebar: a few years ago the scientific consensus was that BPA was so quickly metabolized that unconjugated BPA never entered the general circulation, despite multiple lines of evidence to the contrary. Of course, the studies that conflicted with industry studies were disregarded as being due to laboratory error or contamination. At this point, the science is overwhelming. The current understanding is that either BPA is not detoxified as rapidly as once thought, that there are other routes outside the digestive tract that allow unconjugated BPA in, or the most likely answer being both. Regardless, it's entering the circulation unconjugated and disrupting hormonal signaling)
Righting the ship
So what are you supposed to do? If it's not obvious, one of the first things you need to do is remove sources of inflammation, particularly in the digestive tract as GI inflammation likely contributes to systemic inflammation to a significant degree. This includes removing or limiting things like dairy, gluten, processed seed oils, or any food you are intolerant to.The next thing you can do is limit the amount of fat soluble xenobiotics you are exposed to on a daily basis. BPA is a big one, primarily due to the consumption of food heated in plastic, cooked with plastic utensils, or hot coffee that's exposed to plastic. This includes coffee made in coffeemakers that use plastic including Keurig brewers, coffee that's stored in plastic dispensers at gas stations and convenience stores, and coffee that's consumed through plastic lids. Whether these containers are BPA-free or not is irrelevant, the most frequently used replacement for BPA, BPS, has the same endocrine disrupting effects as BPA and seeps from plastic container in to its contents. Organochlorine pesticides are also a major route of exposure. Other sources of fat soluble toxins include receipts, cosmetics, detergents and perfumes, although these sources shouldn't be ingested but may enter via the skin. :)
One thing you shouldn't do is just start slamming down detoxification promoting supplements and foods. Manipulating the detoxification process to improve your health is a highly individualized process dependent on genetic as well as environmental factors and many substances can have a dramatic impact on the way you feel due to these factors. For example, grapefruit and pomegranate juice are both known to inhibit phase 1 detoxification and can dramatically impact the blood levels of certain drugs, actually a lot of them since they both directly inhibit CYP3A4. So think about that when your slamming down a bottle of Pom. Also, feel free to refer this to your friend who says the human body can easily handle all environmental toxins that are thrown at it when simply drinking grapefruit juice can lead to toxic accumulation of pharmaceutical drugs in a very short amount of time.
For someone on chemotherapy or a drug metabolized via CYP3A4, these products can lead to adverse events. However, for someone like me who is genetically a fast oxidizer, slowing down phase 1 detoxification can reduce the number of products from phase 1 detoxification and allow my phase 2 pathways to catch up, improving fatigue and general well-being due to reduced toxic load.
Another area where Phase 1 inhibitors are useful is people reacting to aflatoxins from mold. Alflatoxins are made more toxic after Phase 1 reactions and can bioaccumulate if Phase 2 is outpaced by Phase 1. Different supplements manipulate detoxification in different ways and this needs to be kept in mind when supplementing to improve the way you feel as well as your overall health.
This brings me to a final point...There are 6 pathways of phase 2 detoxification, don't just focus on methylation and glutathione conjugation. BPA is a great example given that it's conjugated primarily via glucuronidation and sulphation, 2 pathways that use and are induced by different nutrients than methylation and glutathione conjugation. Supporting these two pathways doesn't do too much for the removal of BPA from the body. In fact, simply sweating does more for BPA removal than supporting methylation and glutathione conjugation, although glutathione can act as a back up if the other 2 pathways fail.
Conclusion
I hope this blog has given you a deeper understanding of the detoxification process. If manipulated properly, optimizing your detoxification pathways can transform your health and improve your quality of life. Blindly following health trends without a basic understanding of what you're doing can prevent you from attaining optimal health and could make things worse.Fortunately, the absolute first step is relatively easy to do: reduce inflammation. This is primarily done through lifestyle modification including diet, exercise, stress management, proper sleep, and other lifestyle factors that are associated with inflammation. People with inflammatory conditions such as IBD/IBS, arthritis, or an active autoimmune condition will almost certainly require some other support which can include pharmaceutical drugs, supplements, or a combination of both.
From there, if you want to work your detoxification pathways, the next step is knowing how detoxification works for your specific issues. Having your genome done by 23andme and using promethease or genetic genie to look at your detoxification pathways is very useful and can help you identify dietary and supplemental factors that will save you a lot of time and failure in the long run. It will help you identify which pathways are sluggish and, therefore, which environmental toxins you may be more sensitive to. There are numerous resources on the web that can get you pointed in the right direction.
The final step is to formulate a plan and give it a go, using how you feel as a guide on your success and recording what you do for future reference. Also, keep in mind that initiating any program like this should be discussed with your physician so that any drug-drug and drug-supplement interactions can be avoided.
Thursday, March 31, 2016
3 important ways Vitamin K2 improves gut health
Do you have irritable bowel disease or just overall poor digestion? Do you feel like inflammation runs rampant throughout your body causing you to be tired and achy all day everyday? Are you concerned with your exposure to chemicals such as bisphenol-A(BPA)? Have you been working to fix these problem with dietary changes and supplements with only minor improvement? Well, there may be a new player in the digestive health scene, but it's actually an old player: Vitamin K2.
Vitamin K2 improves gut health in 3 important ways:
1)It helps improve leaky gut by increasing tight junction proteins
2)It reverses inflammation caused by lipopolysaccharide by changing its structure
3)It promotes the 3 phases of cellular detoxification
These 3 powerful steps can dramatically increase your gut health if you have poor Vitamin K2 intake. So let's take a look at Vitamin K2 and its role in maintaining gut health.

At the time, Dr. Price didn't know that Activator X was Vitamin K2, he didn't have the sophisticated equipment to determine what was improving the health of his patients, but he knew where to find it and that it worked synergistically with vitamins A and D by activating proteins that these vitamins helped synthesize. It wasn't until 2008 that Chris Masterjohn was able to put the puzzle together and figure out that Activator X was Vitamin K2 and that animals convert Vitamin K1 in to Vitamin K2 and store it that way.
Once identified as Vitamin K2, research on Activator X started piling up, providing evidence for its positive effect on bone, dental, and cardiovascular health. The story didn't seem to end there. There were some interesting findings on the way that raised some serious questions. The three organs that store the most Vitamin K2 are the pancreas, salivary glands, and brain. None of these organs are directly related to the proposed effects of Vitamin K2 save for the salivary glands and their effect on dental health, so why do they store Vitamin K2? It's role in brain health is still undetermined, but we may have an idea as to why it's found in the pancreas.

There are multiple forms of Vitamin K2 denoted by the letters MK- followed by a number. Humans and all animals store Vitamin K2 as MK-4, and as mentioned before the pancreas stores the most. However, an interesting thing about the MK-4 stored in the pancreas is that it isn't all used in the pancreas, during digestion the pancreas secretes a substantial amount of MK-4 in to the digestive tract via the common bile duct. This led researchers to believe that Vitamin K2 plays a role in synthesizing, secreting, or activating digestive enzymes(2). The exact role of Vitamin K2 in digestion remained elusive, however.
A couple of more recent studied looking at the effect of administration of Vitamins K1 and K2 may have clarified some of these roles. When K1 and K2 were administered to rats, the expression of genes responsible for the synthesis of intestinal alkaline phosphatase(3, 4) and the pregnane X receptor increased(3). Both of these genes play major roles in regulating inflammation and detoxification in the digestive tract, and through different mechanisms.

Many cells throughout the body manufacture alkaline phosphatase, but the primary role of intestinal alkaline phosphatase(IAP) is in gut mucosal defense(5). In the presence of IAP, the cells that form the intestinal lining increase the expression of tight junction proteins. This improves the integrity of the intestinal barrier and prevents the contents of the digestive tract from leaking in to the bloodstream. IAP is also responsible for detoxifying lipopolysaccharide(LPS), a component of the cell wall of gram negative bacteria that induces inflammation.
The human digestive tract contains vast amounts of gram negative bacteria which can cause excessive inflammation if it leaks in to the bloodstream. When cells of the immune system encounter LPS, it is identified as a foreign invader and causes an increase in inflammation. IAP changes the structure of LPS, rendering it undetectable to the immune system and reducing inflammation. IAP is secreted in to the digestive tract throughout the upper small intestine, mixes with the digestive contents, and makes its way through the digestive tract improving tight junction protein expression and detoxifying LPS. This makes IAP an important regulator of the intestinal immune response.

When a toxin binds to the SXR, it induces the production of the phase I detoxification enzyme CYP3A4 which modifies it to make it more water soluble. Next, phase II conjugating enzymes are induced to conjugate the product of phase I, making it readily excretable. Finally, phase III transport is initiated, removing the toxin completely from the cell and removing it from the body in the sweat, urine or feces. In the case of the intestine, SXR essentially chews up and spits the toxin back out in to the intestinal lumen, preventing it from entering the bloodstream where it can cause havoc and sending it out the back door. It performs the same function in the liver which may explain why MK-4 is also stored there.
Given the role of the pancreas in the digestive process and the lack of bioavailability of food-based vitamin K1, it appears as though Vitamin K2 may have another role in human health in addition to its better known role in bone, dental, and cardiovascular health. The role of MK-4 in maintaining gut health may also help explain why supplementing with MK-4 doesn't lead to substantial increases in serum MK-4. Most of it may be taken up by intestinal cells or recycled to the pancreas from the lymphatic system or portal vein.
One more issue that must be mentioned is the human requirement for Vitamin K2. To date, there is no differentiation between Vitamin K1 and K2 in terms of what is considered the adequate dietary intake, there is just a general recommendation for the adequate intake(AI) of Vitamin K. This number is based solely on the amount of Vitamin K required to prevent problems in blood coagulation, it omits the other functions attributed to Vitamin K2 which include bone, dental, cardiovascular, and now gut health. The rate of conversion of K1 to K2 is estimated to be between 5-25%(7), and if the pancreas requires MK-4 for proper digestive function, an AI of total K based solely on coagulation is far from adequate even if you don't consider its function in bone, teeth and blood vessels.

In the functional medicine realm, probiotics, fiber, magnesium, digestive enzymes, and a host of other nutrients and nutraceuticals have attracted a considerable amount of attention and are used therapeutically to improve nutritional deficiencies and overall gut health. One nutrient that hasn't received any attention is Vitamin K2. There is ample evidence to consider Vitamin K2 as a potent modulator of gut health as well as your overall health and well-being.
Vitamin K2 improves gut health in 3 important ways:
1)It helps improve leaky gut by increasing tight junction proteins
2)It reverses inflammation caused by lipopolysaccharide by changing its structure
3)It promotes the 3 phases of cellular detoxification
These 3 powerful steps can dramatically increase your gut health if you have poor Vitamin K2 intake. So let's take a look at Vitamin K2 and its role in maintaining gut health.
Dr. Weston A. Price, DDS
The History of Vitamin K2
Vitamin K2, also known as menaquinone, was first identified by Dr. Weston A. Price in his journey to discover the effects of the modern diet on dental and overall health. In his research, he identified some unknown Activator X found in the butterfat and organs of mammals fed a diet high in grass that had profound effects on dental and overall health. It seemed that this Activator X was somehow responsible for putting calcium where it was supposed to be, in the bones and teeth, while at the same time removing it from places it shouldn't be, the blood vessel walls.At the time, Dr. Price didn't know that Activator X was Vitamin K2, he didn't have the sophisticated equipment to determine what was improving the health of his patients, but he knew where to find it and that it worked synergistically with vitamins A and D by activating proteins that these vitamins helped synthesize. It wasn't until 2008 that Chris Masterjohn was able to put the puzzle together and figure out that Activator X was Vitamin K2 and that animals convert Vitamin K1 in to Vitamin K2 and store it that way.
Once identified as Vitamin K2, research on Activator X started piling up, providing evidence for its positive effect on bone, dental, and cardiovascular health. The story didn't seem to end there. There were some interesting findings on the way that raised some serious questions. The three organs that store the most Vitamin K2 are the pancreas, salivary glands, and brain. None of these organs are directly related to the proposed effects of Vitamin K2 save for the salivary glands and their effect on dental health, so why do they store Vitamin K2? It's role in brain health is still undetermined, but we may have an idea as to why it's found in the pancreas.
The pancreas and its outlet in to the duodeunm
Vitamin K2 in the Pancreas
What purpose does storing vitamin K2 serve in the pancreas? Given the organs function in regulating blood glucose levels, the initial thought was that it had some effect on regulating insulin secretion, and there is evidence that it may have some role there(1). However, the pancreas also plays a pretty significant role in digestion by secreting digestive enzymes in to the gut to help break down food.There are multiple forms of Vitamin K2 denoted by the letters MK- followed by a number. Humans and all animals store Vitamin K2 as MK-4, and as mentioned before the pancreas stores the most. However, an interesting thing about the MK-4 stored in the pancreas is that it isn't all used in the pancreas, during digestion the pancreas secretes a substantial amount of MK-4 in to the digestive tract via the common bile duct. This led researchers to believe that Vitamin K2 plays a role in synthesizing, secreting, or activating digestive enzymes(2). The exact role of Vitamin K2 in digestion remained elusive, however.
A couple of more recent studied looking at the effect of administration of Vitamins K1 and K2 may have clarified some of these roles. When K1 and K2 were administered to rats, the expression of genes responsible for the synthesis of intestinal alkaline phosphatase(3, 4) and the pregnane X receptor increased(3). Both of these genes play major roles in regulating inflammation and detoxification in the digestive tract, and through different mechanisms.
Presence and absence of tight junction proteins in the intestine
Intestinal Alkaline Phosphatase as a Regulator of Gut Mucosal Defense
Many cells throughout the body manufacture alkaline phosphatase, but the primary role of intestinal alkaline phosphatase(IAP) is in gut mucosal defense(5). In the presence of IAP, the cells that form the intestinal lining increase the expression of tight junction proteins. This improves the integrity of the intestinal barrier and prevents the contents of the digestive tract from leaking in to the bloodstream. IAP is also responsible for detoxifying lipopolysaccharide(LPS), a component of the cell wall of gram negative bacteria that induces inflammation.
The human digestive tract contains vast amounts of gram negative bacteria which can cause excessive inflammation if it leaks in to the bloodstream. When cells of the immune system encounter LPS, it is identified as a foreign invader and causes an increase in inflammation. IAP changes the structure of LPS, rendering it undetectable to the immune system and reducing inflammation. IAP is secreted in to the digestive tract throughout the upper small intestine, mixes with the digestive contents, and makes its way through the digestive tract improving tight junction protein expression and detoxifying LPS. This makes IAP an important regulator of the intestinal immune response.
The 3 phases of cellular detoxification
Note: Phase 3 is the excretion process
The Steroid X Receptor and Detoxification
The Steroid and Xenobiotic Receptor(PXR) also plays a major role in gut health, particularly with regard to detoxification. The primary function of the SXR is to bind foreign substances and initiate the 3 phases of cellular detoxification responsible for modifying toxins and removing them from the body. The SXR is responsible for binding chemicals and initiating the metabolism of a variety of toxins, most notable of which is the endocrine disrupting compound bisphenol-A(BPA) ubiquitously found in plastics(6).When a toxin binds to the SXR, it induces the production of the phase I detoxification enzyme CYP3A4 which modifies it to make it more water soluble. Next, phase II conjugating enzymes are induced to conjugate the product of phase I, making it readily excretable. Finally, phase III transport is initiated, removing the toxin completely from the cell and removing it from the body in the sweat, urine or feces. In the case of the intestine, SXR essentially chews up and spits the toxin back out in to the intestinal lumen, preventing it from entering the bloodstream where it can cause havoc and sending it out the back door. It performs the same function in the liver which may explain why MK-4 is also stored there.
Healthy gut=Healthy body
Vitamin K2 as a Regulator of Gut Health
There is ample evidence to point to Vitamin K2 as a regulator of gut health. However, the study identifying its role in upregulation of IAP and SXR expression found both Vitamin K1 and K2 to have these effects in rats. Vitamin K2 is likely of more significance for two reasons. First, the study looked at oral supplementation of K1 and K2 which is vastly different than getting either from food. In both studies, K1 had a greater effect in the upper part of the small intestine, and K1 bound to food may not be very bioavailable there(7). Second, it makes no sense for the pancreas to store and secrete MK-4 in to the duodenum if it isn't serving some purpose in the digestive process, K1 isn't directly involved in this process as far I am aware.Given the role of the pancreas in the digestive process and the lack of bioavailability of food-based vitamin K1, it appears as though Vitamin K2 may have another role in human health in addition to its better known role in bone, dental, and cardiovascular health. The role of MK-4 in maintaining gut health may also help explain why supplementing with MK-4 doesn't lead to substantial increases in serum MK-4. Most of it may be taken up by intestinal cells or recycled to the pancreas from the lymphatic system or portal vein.
One more issue that must be mentioned is the human requirement for Vitamin K2. To date, there is no differentiation between Vitamin K1 and K2 in terms of what is considered the adequate dietary intake, there is just a general recommendation for the adequate intake(AI) of Vitamin K. This number is based solely on the amount of Vitamin K required to prevent problems in blood coagulation, it omits the other functions attributed to Vitamin K2 which include bone, dental, cardiovascular, and now gut health. The rate of conversion of K1 to K2 is estimated to be between 5-25%(7), and if the pancreas requires MK-4 for proper digestive function, an AI of total K based solely on coagulation is far from adequate even if you don't consider its function in bone, teeth and blood vessels.
Gratuitous kitty with a sombrero shot
Conclusion
We are entering a time where the health of the gut is receiving a lot of well-deserved attention for overall health. Not only is human digestive health on the decline, but many of the diseases that we are succumbing to may be partially attributed to poor digestive health. Fixing digestive health is no small task, and our current paradigm of throwing acid blocking medications at basically everything is not working.In the functional medicine realm, probiotics, fiber, magnesium, digestive enzymes, and a host of other nutrients and nutraceuticals have attracted a considerable amount of attention and are used therapeutically to improve nutritional deficiencies and overall gut health. One nutrient that hasn't received any attention is Vitamin K2. There is ample evidence to consider Vitamin K2 as a potent modulator of gut health as well as your overall health and well-being.
Monday, December 14, 2015
5 tips for improving recovery from a concussion
Do you play sports professionally or as a weekend warrior?
Do your children play sports in school or in their free time with friends?
If you answered yes to either of these questions, then you are familiar with the concern over concussions in sports. But what is a concussion?
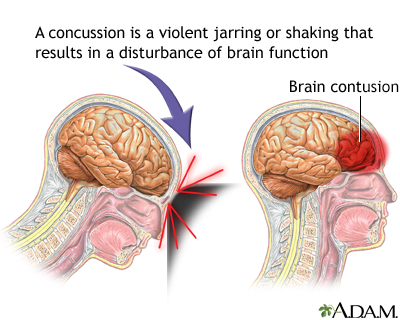
While traumatic brain injuries can lead to concussions, not all do. Light blows to the head are typically not a concern and are common. But when these blows become strong enough to damage the brain and alter brain function, they're referred to as concussions.
Complete recovery from a concussion depends on many things including the severity of your injury, your current health, and if you have recovered from a previous brain injury.
Chances are the increased awareness brought about by concussions in the NFL and combat sports has you worried about the long term effects of repeated blows to the head. While most people who suffer from a sports-related concussion recover within 7-10 days, around 10% can experience symptoms beyond 10 weeks. Some can experience symptoms for years afterwards.
An issue that makes this worse is that sometimes the symptoms of a concussion are not obvious. This is a major problem because suffering another blow to the head before the brain has fully recovered can lead to long-term problems and even death as increased swelling can cut off blood supply to the brain.

As you can see, it's important to make sure you heal properly from a concussion. While some of the factors are out of your control, there are things you can do to speed up the process and make sure the damage heals completely.
So let's go over some tips on how to improve recovery from a concussion. We'll go over some of the basics in the tips in this blog. For a more detailed description of why each tip is important, take a look at this blog on the science behind concussions.

Tip 1: Avoid alcohol
This tip may seem like a no-brainer, but the importance of avoiding alcohol can't be overstated. Even if you're feeling fine symptom-wise, alcohol can delay recovery from a concussion and prevent full recovery.Another issue with drinking alcohol during recovery from a concussion is that it increases your risk for a second concussion. It can do this by delaying recovery or by negatively affecting balance and coordination, which are already a problem for people recovering from a concussion. In fact, the effects of alcohol are often amplified in people recovering from a concussion.
The main way that alcohol affects recovery from a concussion is through inflammation. Alcohol increases inflammation throughout the body, something you'll want to avoid while recovering from a concussion. Chronic inflammation causes immune cells in the brain to kill healthy brain cells and prevent damaged ones from healing.
Chronic traumatic encephalopathy(CTE) is the disease affecting former NFL players that has brought all of these concussion issues to light. Alcohol abuse appears to be a major player in this disease in two ways.
As mentioned before, alcohol abuse delays recovery from concussion. This disease is thought to be the consequence of repeated blows to the head. These repeated concussions may lead to an accumulation of damage over the years that never heals with chronic alcohol use.
One of the hallmark symptoms of CTE is changes in mood and personality. Since the disease also lowers your inhibitions, it increases the likelihood that you drink, a double-edged sword.
Bottom line, don't drink alcohol while recovering from a concussion. If you choose to drink alcohol and have a history of concussions, do so in moderation and take other steps to prevent further damage.

Tip 2: Eat fish and other foods high in Omega 3 fatty acids
Omega 3 fatty acids are found in fatty fish, nuts, eggs, and pasture-raised animals. Omega 3 fatty acids are important for proper brain function, but they also have very specific benefits for people recovering from a concussion.DHA, one of the omega 3 fatty acids, is necessary for proper brain development and to maintain function throughout adulthood. It's a major component of brain cells because it's necessary for the production of myelin, a coating that covers nerve cells to speed nerve transmission. In fact, the brain contains the most DHA in the body.
DHA also has other positive effects in the brain. It promotes neurogenesis, the formation of new neurons throughout life. Neurogenesis is important for forming new memories and healing damage to the brain.
This is interesting because many people who do not fully recover from traumatic brain injury have impaired neurogenesis. This can lead to mood problems which are helped with DHA.
One final benefit of omega 3 fatty acids is that they lower inflammation throughout the body. They not only lower inflammation directly, they help reduce levels of lipopolysaccharide(LPS). LPS causes more inflammation and prevents recovery from concussion by causing more damage.
The bottom line here is to eat fish regularly, but don't overdo it. Larger fish contain high levels of mercury which is toxic. Eating smaller fish such as sardines or anchovies regularly can give you some Omega 3 fatty acids while limiting your exposure to mercury.

Tip 3: Eat lots of colorful fruits and vegetables
Fruits and vegetables have many components that can help you recover from a concussion. Many polyphenols such as the curcumin found in turmeric, the sulforaphane found in broccoli and brussel sprouts, and the pterostlibene found in blueberries reduce inflammation AND promote neurogenesis at the same time.These are just a few of the many polyphenols that can improve recovery from a concussion. And the best part is, polyphenols are what give fruits and vegetables their color. It's easy to identify vegetables that are high in polyphenols, they're the most colorful.
Fruits and vegetables are also very nutrient dense. This means that they are rich in vitamins and minerals. These vitamins and minerals are essential to heal damage to the brain.
Another component of fruits and vegetables that improves recovery from a concussion is fiber. Fiber helps calm inflammation by reducing the amount of LPS that enters the bloodstream from the gut. It also helps seal off the blood-brain barrier so that any LPS that enters the bloodstream can't access the brain.
When it comes to fruits and vegetables you should eat at least 5 different colors of fruits and vegetables a day. It's also important to eat at least 6 cups of vegetables every day. Get your veggies from a variety of categories including cruciferous vegetables, leafy greens, and root vegetables.

Tip 4: Make sure you get enough thiamine
Thiamine, also called vitamin B1, is necessary for proper brain function. Thiamine is required for neurons to generate energy. Not enough thiamine can kill neurons and cause high levels of inflammation.Since thiamine is so crucial for the brain, most of the signs of thiamine deficiency are related to brain function. This includes depression, confusion, poor memory, and balance/coordination problems. These symptoms are also common when a concussion isn't healing properly.
While foods that are high in thiamine are important, supplements can be more effective because you can absorb them better. Fat-soluble forms of thiamine such as sulbutiamine have been shown to create much higher levels of thiamine in the brain than the water soluble kinds found in multi-vitamins.

Tip 5: Get out of bed and exercise...gradually
Over the years, strict bed rest has been the go-to therapy for a concussion. However, recent scientific evidence is showing that prolonged bed rest may actually delay recovery from a concussion.A recent study found that student athletes who remained on bed rest for 2-5 days after a concussion reported more post-concussion symptoms than athletes who restarted their normal routine within 1-2 days. The athletes on longer bed rest also reported that it took longer for their symptoms to go away.
Another study followed concussed athletes for 12 weeks. One group just stretched while the other performed 20 minutes of aerobic exercise for 20 minutes a day, 6 days a week. What makes this study cool is that they looked at brain activity in each of the 2 groups and compared it to people who did not experience a concussion.
The results of this study were incredible. Athletes who performed aerobic exercise saw better improvements in brain activity than those who just stretched. In fact, their brain activity patterns were not different from people who hadn't experienced a concussion.
Why would aerobic exercise improve recovery from a concussion? It's because aerobic exercise increases neurogenesis. Aerobic exercise also has the added benefit of reducing chronic inflammation.
Unlike the other 4 tips that can be implemented immediately, exercise must be added gradually. A good rule of thumb is to let your symptoms dictate when you begin aerobic exercise. Your symptoms should also dictate the intensity of your exercise.
Start with walking and only progress to more intense aerobic exercise if you don't experience symptoms. Symptoms to look for include headache, dizziness, or confusion. Once symptoms allow, increase exercise intensity so that your heart rate is in the 60-75% of maximum heart rate range. Stop exercise if symptoms worsen.
Conclusion
As we learn more about the effects of concussions, treatment options will improve. Currently, the only treatment option is rest, observation, and pain relievers as needed. Very little attention is given to lifestyle factors other than rest.Diet and exercise are 2 lifestyle factors that can improve recovery from concussion. Both factors improve recovery by reducing inflammation and increasing neurogenesis.
On the diet side of things it's important to avoid alcohol, eat fish, eat your fruits and veggies, and get enough thiamine to support brain recovery. These 4 diet tips can be implemented immediately following a concussion since there is no risk.
Exercise, on the other hand, must be approached with more caution. Full bed rest for a day or two following a concussion may be necessary if low level activities worsen symptoms. You can begin walking as symptoms get better and add in more intense aerobic exercise as symptoms allow.
These 5 tips will improve recovery from a concussion. When paired with monitoring by your healthcare professional, the risk to implementing these tips is negligible. But, you should discuss these tips with your doctor before beginning.
Looking for more information on what you can do to improve recovery from a concussion? We will be expanding these tips in the future. Like us on facebook for updates on this article and future articles on the topic.
Friday, November 20, 2015
Conquering concussions: Optimizing recovery following traumatic brain injury
Concussion due to traumatic brain injury(TBI) is a relatively common occurrence that mostly happens to young men and children due to falls, motor vehicle accidents, or contact sports. They are also common in battle exposed military personnel. A concussion is defined as a traumatic brain injury that alters the way the brain functions. One of the major problems with concussions is that they can have acute and long-term effects. While the vast majority of concussions tend to be mild and people fully recover within 1 to 6 weeks, around 15% may experience persistent, long term effects on brain function. These long term effects include headaches, mood and anxiety disorders, dizziness and motor problems, problems with memory, and more. When someone experiences these symptoms for longer than six weeks, they are said to have post-concussion syndrome. It is estimated that 5.3 million Americans are living with disability due to traumatic brain injury(1).
Persistent symptoms following a concussion are likely due to an impaired ability to heal the damage from the traumatic incident. This can be due to the severity of the trauma as well as inter-individual variability in the ability to heal the damage. Within the inter-individual variability aspect, we have genetic as well as environmental influences. Since we have no control over genetic differences, it becomes important to look at environmental factors and how we can manipulate them. When we dig in to some of the physiological effects of traumatic brain injury, a few lifestyle factors stick out as potential environmental influences that can be manipulated successfully.
Traumatic brain injury and blood brain/intestinal barrier integrity
Within a few hours after a traumatic brain injury, integrity of the blood brain barrier is lost depending on multiple factors including the severity of the injury(2, 3, 4). While this disruption is typically resolved within days to weeks, it can sometimes remain disrupted for months or even years after the injury(4). The blood brain barrier functions to separate the circulation within the central nervous system from the general circulation. Certain substances within the general circulation are necessary for optimal brain function while others are neurotoxic and must be kept out of the central nervous system. The blood brain barrier functions to prevent neurotoxic substances from crossing in to the central nervous system and causing destruction of brain tissue. Obviously a disrupted blood brain barrier is less than ideal when healing from traumatic brain injury, particularly if it remains disrupted for a long period of time.While the damage related to traumatic brain injury is typically thought of as occurring solely in the brain, TBI also causes dysfunction of the gastrointestinal tract including intestinal barrier dysfunction and disturbed motility(5, 6). This is likely due to the local effects of the traumatic brain injury causing autonomic dysregulation, potentially through the vagus nerve. The vagus nerve links the brain and gut by providing a conduit through which the 2 organs can communicate. Damage to the brain may derail this communication system and cause gastrointestinal dysfunction. A study looking at the vagus nerve in mice found that vagus nerve stimulation attenuated the increase in intestinal permeability caused by traumatic brain injury(7).
Adding another layer of complexity to the situation is that research has shown that bacteria found in the digestive tract that help regulate the permeability of the intestinal barrier also appear to regulate the blood brain barrier(8). This suggests that one environmental influence that may dictate the ability to recover from TBI is the composition of bacteria in the gut. This may not only have to do with regulating intestinal barrier integrity, it may have just as much to do with regulating blood brain barrier integrity through the vagus nerve.
Immunoexitotoxicity and recovery from traumatic brain injury
Intestinal barrier dysfunction may not seem like a big issue in recovering from traumatic brain injury, but it is probably the most important thing that needs to be addressed aside from avoiding a recurrent injury before the damage is repaired. Intestinal permeability can prolong and even prevent recovery from traumatic brain injury as it causes an over-activation of the brain’s immune system that, when chronically active, is responsible for causing more damage than the actual injury itself, a process termed immunoexcitotoxicity(9). If this process is allowed to continue, complete recovery is unlikely.The primary issue with intestinal permeability is that lipopolysaccharide(LPS), a component of the cell wall of gram-negative bacteria found in the digestive tract, is harmless in the digestive tract but toxic in the blood circulation. If the intestinal barrier is compromised, LPS can leak from the digestive tract in to the circulation. Furthermore, LPS can hitch a ride in to the lymphatic system on chylomicrons, the extent of which is determined by the amount and types of fat in the diet. LPS causes excessive immune system activation throughout the body. Compounding the issue is that TBI also causes permeability of the blood brain barrier, which should separate the circulation in the brain from that of the rest of the body. This allows LPS and inflammatory cytokines that are in the general circulation to enter the central nervous system where they are neurotoxic.
Glutamate, the most abundant neurotransmitter in the brain, has many important roles there. However, it does not play well with LPS. Too much glutamate can be toxic, but when subtoxic levels of glutamate are exposed to subtoxic levels of LPS or inflammatory cytokines, resident immune cells of the brain called microglia begin destroying neurons and secreting more glutamate causing a vicious cycle, the aforementioned immunoexcitotoxicity(9). This process prevents optimal recovery from TBI, underscoring the importance of restoring intestinal barrier integrity. Immunoexcitotoxicity is thought to underlie chronic traumatic encephalopathy, the degenerative neurological condition seen in retired NFL players as well as combat veterans and athletes. It also plays a role in Alzheimer's disease which is why both CTE and Alzheimer's have essentially the same symptoms.
Neurogenesis and recovery from traumatic brain injury
During recovery from traumatic brain injury, and even throughout life, new neurons are created through a process known as neurogenesis. This process promotes learning as well as healing from traumatic brain injury and is partially regulated by microglia. When microglia are in their resting, ramified state they appear to be an integral player in promoting neurogenesis(10). When microglia remain in a primed, activated state due to inflammation or LPS in the local environment, the research shows they inhibit neurogenesis and destroy healthy neurons(11, 12) while disrupting the blood brain barrier(13). This same process occurs in the hippocampus during chronic intestinal inflammation(14) which both causes and is a consequence of a disrupted intestinal barrier. This allows LPS to leak from a disrupted intestinal barrier in to the bloodstream and from the bloodstream across a disrupted blood brain barrier in to the central nervous system where it can interact with microglia. The combination of impaired neurogenesis coupled with hyperactive microglia that destroy neurons is likely a significant factor in delayed healing from TBI.The enteric nervous system
As mentioned above, stimulation of the vagus nerve can attenuate intestinal barrier dysfunction due to TBI. Yoga, diaphragmatic breathing, meditation, and other relaxation techniques are all ways to stimulate the vagus nerve. However, vagus nerve stimulation is not the only way to improve intestinal barrier function. While the brain exerts a significant amount of influence on the function of the digestive tract, the digestive tract has its own nervous system, the enteric nervous system, which can function on its own without help from the brain. Studies have shown that despite severing the vagus nerve, the enteric nervous system still functions via reflexive activity(15).The enteric nervous system appears to function as a back-up generator to the digestive system capable of holding intestinal barrier integrity while the brain heals from injury. While the blunt force trauma that occurs during traumatic brain injury may disrupt the communication of the gut-brain axis, the enteric nervous system can maintain intestinal barrier integrity until the vagus nerve comes back online. Unfortunately, the standard American diet is fairly devoid of the nutrients necessary to keep this generator going and an individual's diet prior to the traumatic brain injury is probably a significant factor in how likely that person is to fully recover from it. This all comes back to the composition of bacteria in the gut which are, to a great degree, dictated by diet. This does not mean all is lost as certain dietary, behavioral, and exercise interventions implemented after a traumatic brain injury can optimize recovery and potentially prevent chronic symptoms.
Functional goals in healing from traumatic brain in jury
The functional goals with diet after traumatic brain injury are to maintain intestinal barrier integrity to prevent LPS from entering the circulation and activating microglia while providing nutrients that give the brain what it needs to heal. The great part about diet is that it is an intervention that can be implemented immediately. In addition, relaxation methods such as yoga and meditation can be used to restore function of the vagus nerve and restore communication between the brain and gut. Finally, and this may seem counterintuitive, but aerobic exercise can be initiated as symptoms allow. While the standard of care for concussions has always been lots of bed rest, recent research is showing long term bed rest may not be optimal and is possibly destructive.In healthy individuals, aerobic exercise has been shown to improve autonomic function(16, 17) and promote neurogenesis in the hippocampus(18). A recent review of the literature cites ample evidence that aerobic exercise is a potent intervention for improving recovery from traumatic brain injury through improved autonomic function, enhanced neurogenesis, and both reduced inflammation and brain immunoexitotoxicity. This means that we have direct control over improving intestinal barrier function and neurogenesis and, thus, may have significant control over recovery from TBI.
Conclusion
In the initial stages of TBI, eating the proper diet will keep the back-up generator going while a combination of yoga or meditation and aerobic exercise, when tolerable, can help to expedite recovery by repairing the damage to the autonomic nervous and re-establishing the brain-gut connection by improving function of the vagus nerve.Thursday, October 29, 2015
Methylation and the folate cycle: You're on the wrong path(way)!

The picture above represents the methylation cycle and the pathways that help power it. On the right side in grey you have a partial representation of the folate cycle while on the left side we have a mostly complete representation of choline metabolism. Both of these processes intersect the methylation cycle at the same point, a critical point where homocysteine is recycled in to methionine. High homocysteine levels are a bad scene as homocysteine is toxic, so having efficient conversion of homocysteine to methionine is important to prevent this scenario.
The folate cycle and choline metabolism balance each other out
Many people are aware of how the folate cycle impacts methylation, many are even aware of gene polymorphisms they have that negatively impact this point of the methylation cycle. However, few realize that having a defect on one side is typically compensated for by the other side. In other words, when the folate cycle doesn't run well, choline metabolism can pick up the slack with respect to the methylation cycle and convert homocysteine to methionine, and there are studies that show this(1, 2). It's important to understand the opposite to be true as well, when choline metabolism doesn't work well the folate cycle can pick up the slack with regard to the methylation cycle.The problem is that most people with polymorphisms related to the folate cycle focus on getting more folate or increasing methyl-B12 in the hopes of improving methylation. This is a fool's errand as the primary polymorphism occurs in the MTHFR gene, which is the rate limiting step and occurs prior to the step in the cycle where homocysteine is recycled to methionine. This isn't to say that you shouldn't get adequate B12 and folate, just that this pathway will always be inefficient at recycling homocysteine to methionine. Why focus on it then?
Dietary choline vs. choline synthesis

Choline is not considered an essential nutrient in the United States by definition because people who get adequate methionine can make it and people with adequate folate intake need less of it. The problem with this definition is that inadequate choline increases the demand for both methionine and folate and it does not take into consideration polymorphisms, such as MTHFR, that increase choline requirement. This caused the Institute of Medicine to reclassify choline as an essential nutrient.
The recommended intake for choline is set at 425mg/day for women and 550mg/day for men, but people vary in their need for choline. People with MTHFR polymorphisms as well as pregnant women need higher intakes of choline. Foods that are high in choline include eggs, oysters, fish, poultry, beef liver, cruciferous vegetables and peanuts. Supplementing choline in the form of soy or egg lectithin, choline bitartrate, or phosphatidylcholine is also effective and well tolerated.
Signs you need more choline
One of the easiest signs that you need more choline is that you are a human, living in the United States. Only 10% of older children and adults get adequate choline in their diet and not all show overt clinical symptoms(3). Fatty liver and muscle damage are common, but these symptoms tend to be subclinical in nature so most people would have no idea they are experiencing this until something significant happens. I would suspect that most symptoms of choline deficiency would primarily center around functions that rely on methylation or the neurotransmitter acetylcholine. Since most people who are currently trying to improve the function of the folate cycle are familiar with symptoms associated with poor methylation, I'll focus on the symptoms associated with low acetylcholine.Acetylcholine is formed when the enzyme choline acetyltransferase turns acetyl CoA and choline in to acetylcholine and coenzyme A.

Many executive functions such as sleep, stress, digestion, GI motility, and memory all rely on sufficient acetylcholine. In regard to sleep, acetylcholine is very important for REM sleep, the stage of sleep where dreams, and potentially memory consolidation, occur. Within the autonomic nervous system, preganglionic nerves of both the sympathetic and parasympathetic branches use acetylcholine as well as postganglionic nerves of the parasympathetic branch. Symptoms of acetylcholine deficiency in the autonomic nervous system would likely include anxiety and mood disorders.
During digestion, acetylcholine is responsible for the secretion of enzymes in the stomach as well as motility throughout the digestive tract. Impairments in these functions can lead to IBS, SIBO, or poor nutrient status due to problems in absorption. An interesting side note is that both caffeine and nicotine, which tend to increase GI motility, work by increasing acetylcholine levels or activating acetylcholine receptors. Caffeine works by inhibiting the breakdown of acetylcholine and nicotine works as a substitute for acetylcholine by stimulating nicotinic acetylcholine receptors. Linking digestion and the autonomic nervous system together, the primary neurotransmitter used by the vagus nerve is acetylcholine. The vagus nerve is known as the conduit through which the brain and gut communicate with one another.
Finally, with regard to memory, acetylcholine is very important for the formation of new memories and deficiency can impair working memory(4). This would most likely present as brain fog. Further underscoring the importance of acetylcholine for memory, the class of drugs most used for Alzheimer's disease work by inhibiting the same enzyme that coffee inhibits and people with Alzheimer's disease produce less acetylcholine and have dysfunctional nicotinic acetylcholine receptors(5).
So, it's as simple as supplementing with choline, right? If only it were that simple...
Subscribe to:
Posts (Atom)